

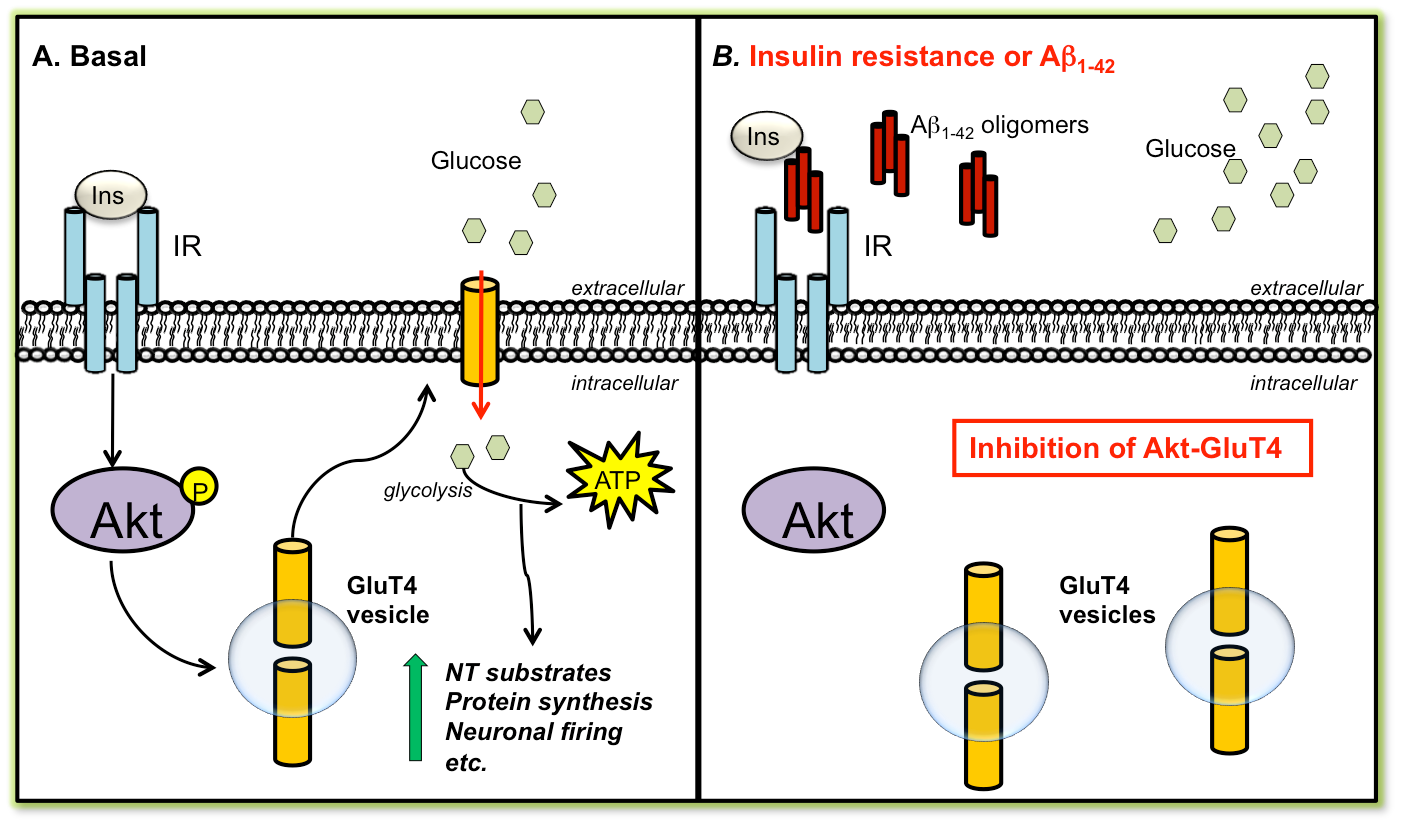
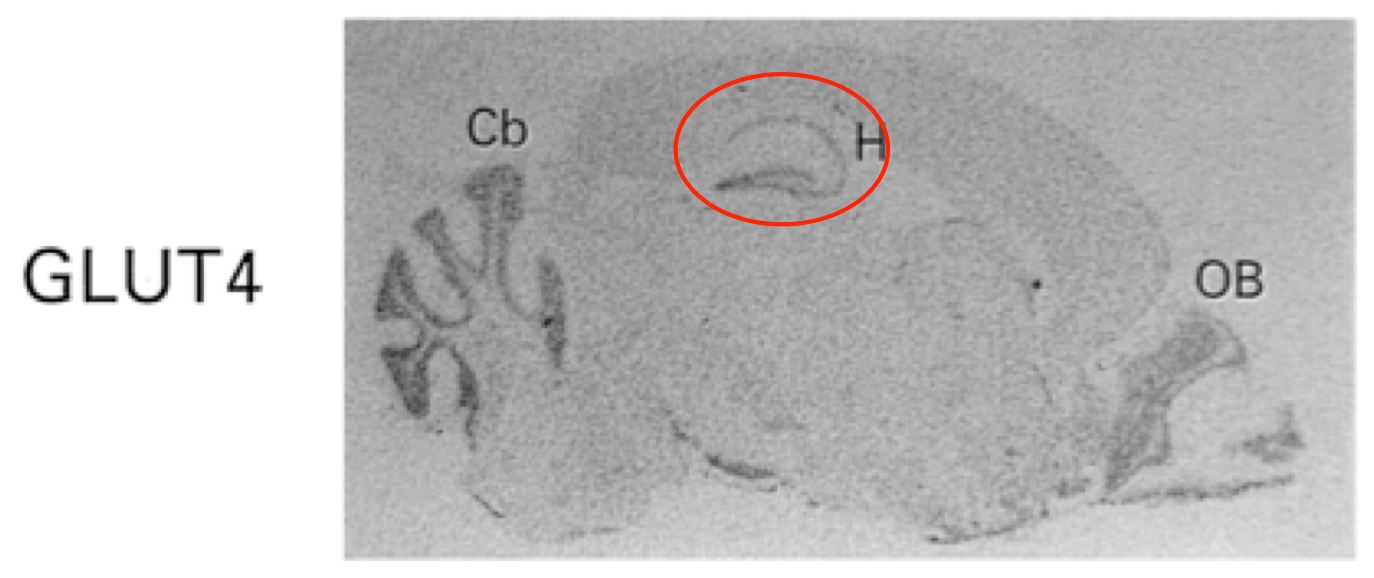
Under the leadership of principal investigator Dr. Ewan McNay, the lab conducts research on brain metabolism and metabolic control of cognitive processes, with particular emphases on the neural impacts of diabetes, insulin, Alzheimer's disease, and glucose.
Resources
Past Collaborations
McNay Lab
Ewan McNay
1400 Washington Avenue
Albany, NY 12222
United States